基于高通量测序的宏基因组学技术,已成为研究微生物群落组成、结构及功能最主要的技术手段。宏基因组研究通常采用16S rRNA测序以获得物种谱信息,或采用全基因组随机测序WGS以得到功能基因谱信息,或两种策略同时采用。但是受测序技术和实验方法本身的限制(即短序列和小片段文库),这些研究会割裂物种谱和功能谱之间的联系。这是因为16S rRNA序列在宏基因组拼接时被视为重复序列,或被拼接到一起,或被舍弃,无法建立其与侧翼的蛋白编码基因的连接,导致16S rRNA物种谱信息与功能基因谱信息的割裂。这给环境微生物物种多样性(尤其是种下多态性)和功能多样性的研究带来严重障碍。
近来,中科院北京生科院赵方庆研究团队在现有宏基因组学技术的基础上,提出一种全新的宏基因组研究策略,即16S rRNA-侧翼序列环化测序及计算技术(RiboFR-Seq,ribosomal RNA gene flanking region sequencing)。通过该技术,可以同时获得16S rRNA V4/6高变区及16S rRNA上游的蛋白编码基因序列。基于此数据,能够建立起16S rRNA与宏基因组拼接序列的物理关联,校正或补充彼此注释的结果,实现准确无偏的宏基因组数据解析,进而快速、准确和全面地解析环境样品中微生物的组成和功能。
研究人员利用该技术,进一步对人体共生微生物和海洋生物表面附生微生物群落开展了研究。从实际数据分析结果来看,RiboFR-seq方法可以实现对宏基因组中16S rRNA拷贝数的测定,从而修正了由于16S rRNA拷贝数差异导致的菌群丰度估计偏差,所得到的菌群组成更能反映环境中的真实情况。此外,利用“桥连序列”信息,对16S扩增子和全基因组测序拼接结果进行重新注释,可辅助宏基因组数据的拼接和组装。本技术首次建立了宏基因组中物种谱和功能基因谱的有效关联,为宏基因组学研究尤其是未知环境条件下微生物组的研究,提供了全新的思路和方法。
本工作由中科院北京生科院赵方庆研究团队的博士后张延明和博士研究生冀培丰共同完成,目前已在国际学术期刊Nucleic Acids Research在线发表。本研究得到国家自然科学基金项目的资助。
论文链接
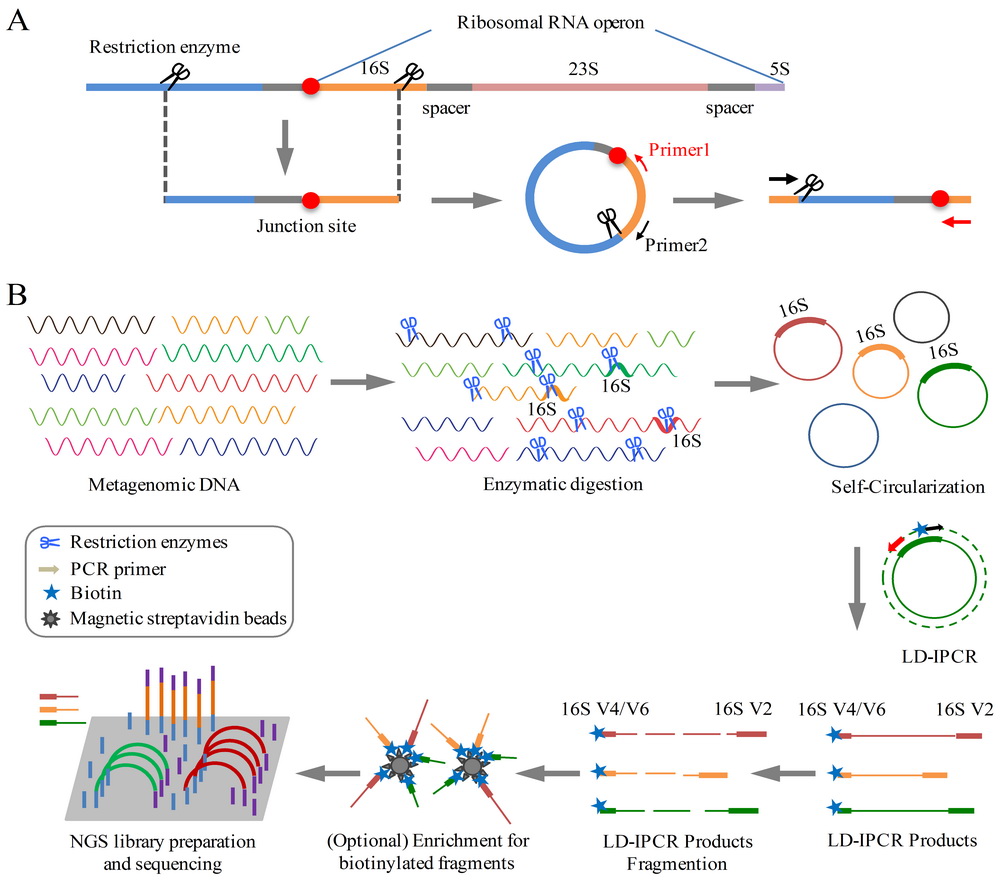
图1. RiboFR-seq的技术原理
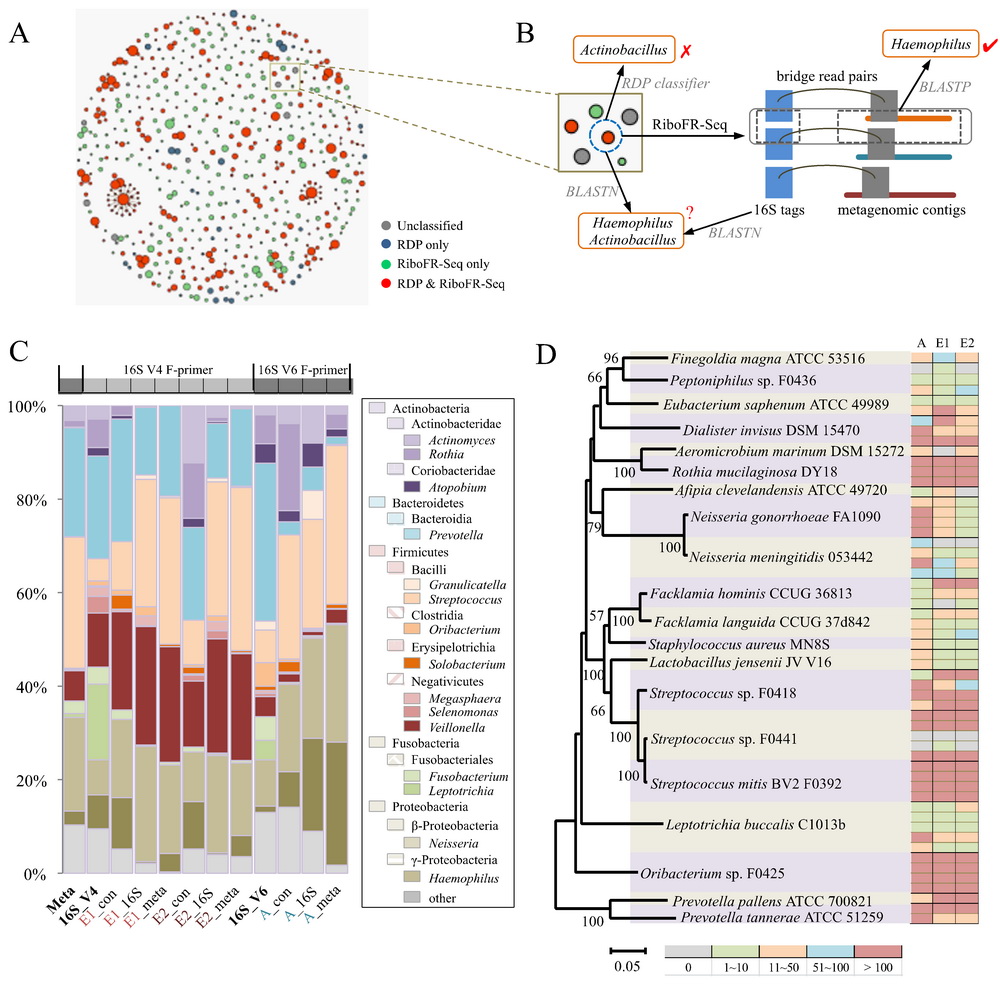
图2. RiboFR-seq技术在口腔微生物组中的应用

